Retour au format normal
5.9 Hypokaliémie
(Q 219)16 juin 2001
par
Objectifs :
Savoir définir l’hypokaliémie et reconnaître les principales complications de l’hypokaliémie
L’hypokaliémie est définie par une concentration plasmatique de potassium inférieure à 3,5 mmol/l. L’hypokaliémie est l’une des anomalies hydroélectrolytiques les plus fréquemment rencontrées en médecine clinique [1].
1. Signes et complications de l’hypokaliémie
1.1. L’hypokaliémie est souvent asymptomatique
La plupart des patients avec une hypokaliémie chronique sont asymptomatiques et la concentration plasmatique de potassium abaissée est découverte par hasard sur un examen sanguin systématique.
1.2. L’hypokaliémie peut induire des signes cliniques ou des complications dont la gravité est liée au degré de l’hypokaliémie et/ou à la rapidité de constitution.
Les hypokaliémies < 3.0 mmol/l et/ou aiguës peuvent être responsables de :
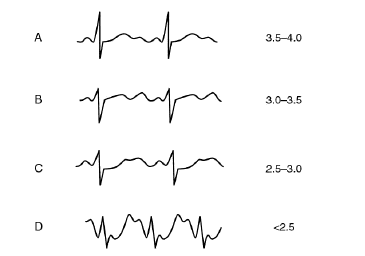
Signes ECG de l’hypokaliémie
- L’hypokaliémie peut favoriser l’augmentation de pression artérielle. L’effet antihypertenseur des diurétiques thiazidiques est diminué par l’hypokaliémie et majoré par la réplétion potassique [4].
- L’hypokaliémie altère la libération d’insuline et la sensibilité à l’insuline des organes-cibles ce qui aggrave l’hyperglycémie chez les patients diabétiques ou peut favoriser la survenue d’un diabète chez les sujets prédisposés.
- Une hypokaliémie entraîne souvent une polyurie modérée de 2 à 3 litres par jour liée à la fois à une augmentation de la soif et à un diabète insipide néphrogénique modéré.
- L’hypokaliémie chronique et sévère en association avec un hyperaldostéronisme peut aboutir à la formation de kystes rénaux [5].
2. Causes des hypokaliémies
2.1. Carence d’apport
La carence d’apport est une cause exceptionnelle d’hypokaliémie [6]. Une hypokaliémie est possible dans les deux circonstances suivantes mais doit toujours faire rechercher une cause supplémentaire de perte potassique :
2.2. Hypokaliémie de redistribution (ou transfert)
2.2.1. Alcaloses
L’alcalose, métabolique ou respiratoire, favorise l’entrée de potassium dans les cellules. Cet effet direct de l’élévation du pH extracellulaire est relativement modéré avec une baisse de la kaliémie de - 0,4 mmol/l pour chaque augmentation de 0,1 du pH sanguin. Cependant l’hypokaliémie est très souvent associée à l’alcalose métabolique probablement parce que le mécanisme de la maladie sous-jacente (diurétiques, vomissements, hyperaldostéronisme) induit à la fois une perte urinaire de protons et de potassium.
2.2.2. Hyperinsulinisme
L’insuline fait rentrer le potassium dans les cellules musculaires squelettiques et les cellules hépatiques. Cet effet est particulièrement marqué avec l’administration d’insuline chez des patients en acidocétose diabétique ou avec une hyperglycémie importante sans cétose.
2.2.3. Hyperactivité béta-adrénergique
Les catécholamines agissant via les récepteurs béta-2-adrénergiques stimulent la captation cellulaire de potassium. Une hypokaliémie transitoire peut être observée dans toute situation s’accompagnant d’une hyperadrénergie induite par le stress, comme au cours des maladies aiguës, de l’ischémie coronaire ou de l’intoxication par la théophylline. De même l’administration d’agonistes béta-adrénergiques tels que l’albutérol, la terbutaline ou la dopamine utilisés pour traiter l’asthme, l’insuffisance cardiaque ou prévenir un accouchement prématuré peuvent induire une baisse de la kaliémie de 0,5 à 1 mmol/l. Cet effet hypokaliémiant de l’adrénaline peut être bloqué par un bétabloqueur non sélectif mais inversement un bétabloqueur sélectif béta-1 ne confère aucune protection.
2.2.4. Paralysie périodique hypokaliémique
Cette maladie rarissime est caractérisée par des épisodes de paralysie ou de faiblesse musculaire pouvant toucher les muscles respiratoires et donc avec un risque léthal potentiel. Au cours des poussées aiguës déclenchées par le repos après un exercice, le stress ou un repas riche en glucides, la kaliémie peut descendre jusqu’à 1,5 à 2,5 mmol/l. La paralysie périodique hypokaliémique peut être soit génétique, soit acquise.
2.2.5. Anabolisme cellulaire
L’anabolisme cellulaire intense, notamment des cellules hépatopoïétiques, s’accompagne d’une captation de potassium et d’une hypokaliémie. Cette circonstance est plus fréquemment observée au cours de l’administration de vitamine B12 ou d’acide folique pour traiter une anémie mégaloblastique ou de l’administration de GMCSF pour traiter une neutropénie.
2.2.6. Intoxications par le baryum ou par la chloroquine
Ces intoxications peuvent causer une hypokaliémie sévère inférieure à 2 mmol/l pour la chloroquine. Ces médicaments bloquent les canaux potassium de la membrane cellulaire.
2.3. Augmentation des pertes intestinales de potassium
La concentration de potassium dans les liquides digestifs bas est relativement élevée, 20 à 50 mmol/l. Les pertes digestives d’origine basse (diarrhées, laxatifs, drainage digestif) sont cause d’hypokaliémie [7]. L’origine digestive de l’hypokaliémie rend compte d’une kaliurèse et d’un GTTK abaissés.
Le liquide de diarrhée est riche à la fois en bicarbonate et en potassium, ce qui est responsable habituellement d’une acidose métabolique tout du moins en aigu.
Chez les patients ayant des diarrhées induites ou en cas d’abus de laxatifs plus ou moins occulte [8] ou chez les patients ayant des diarrhées prolongées sur des périodes importantes par exemple à la suite d’un adénome villeux ou d’une tumeur sécrétant du VIP (vipome), l’acidose métabolique peut être au contraire remplacée par une alcalose métabolique hypokaliémique. Dans ce cas, l’alcalose est attribuée à la contraction volémique induite par les déplétions prolongées.
2.4. Hypokaliémie par fuite urinaire
2.4.1. L’association d’une hypokaliémie avec fuite urinaire de potassium et hypertension artérielle doit faire évoquer en priorité un hyperminéralocorticisme.
Sur une base clinique, trois sous-groupes d’hyperminéralocorticisme peuvent être schématiquement distingués selon le niveau de rénine et d’aldostérone circulante.
Il s’agit des syndromes adrénogénitaux (hypersécrétion de désoxycorticostérone) et des syndromes de Cushing (hypersécrétion de cortisol), de toutes les situations s’accompagnant d’une diminution d’activité de la 11-béta-hydroxystéroïde déshydrogénase (syndrome d’excès apparent en minéralocorticoïde). La diminution de l’activité 11-béta-HSD peut être soit génétique soit acquise notamment en présence d’un inhibiteur comme l’acide glycérrhétinique contenu dans la réglisse, le zan, l’antésite ou en présence d’un fort excès de cortisol endogène circulant (Cushing tumoraux).
On en rapproche le syndrome de Liddle (ou "pseudo-hyperaldostéronisme"), une maladie génétique rare, à transmission autosomique dominante, liée à une mutation d’une sous-unité du canal épithélial sodium dans le tube cortical (ce canal est l’une des principales protéines cibles de l’aldostérone dans cette cellule). La mutation est dite "activatrice" car elle simule une hyperactivité de l’aldostérone en augmentant la réabsorption de sodium en échange de la sécrétion de potassium et de proton.
2.4.2. Hypokaliémie avec fuite urinaire de potassium sans hypertension ou tendance à l’hypotension
L’hypomagnésémie explique l’hypokaliémie associée à différentes tubulopathies, notamment induites par les aminoglycosides ou par l’amphotéricine B. L’hypokaliémie survient chez la moitié des patients traités par amphotéricine B. Celle-ci interagit avec les stéroïdes membranaires favorisant l’augmentation de perméabilité et la sécrétion de potassium à travers la membrane luminale.
Ceci est plus souvent observé au cours des traitements diurétiques notamment par les inhibiteurs de l’anhydrase carbonique (acétazolamide, Diamox®), les diurétiques de l’anse (furosémide, Lasilix® ; bumétamide, Burinex®) et les diurétiques agissant dans le tube contourné distal (diurétiques thiazidiques).
On peut rapprocher certaines maladies génétiques rares associant une alcalose métabolique avec contraction volémique, hypokaliémie avec fuite rénale et liées à des anomalies constitutives des protéines assurant le transport vectoriel du sodium dans le néphron distal.
Le syndrome de Bartter résulte d’une mutation inactivatrice sur l’un des 3 gènes codant pour NaK2Cl, ROMK ou ClCNKB, 3 protéines de transport membranaire du Na ou du K dans la branche ascendante large de l’anse de Henle. Le tableau est celui d’une intoxication par un diurétique de l’anse de présentation néonatale ou précoce. Le risque est lié aux épisodes de déshydratation extracelullaire et à l’insuffisance rénale secondaire à l’hypercalciurie et à la néphrocalcinose.
Le syndrome de Gitelman est une variante correspondant à une mutation inactivatrice sur le gène codant pour NCCT, la protéine de co-transport NaCl au niveau du tube distal. La présentation est celle d’une intoxication par diurétiques thiazidiques et associe une hypocalciurie, une hypomagnésémie et une présentation à un âge plus tardif.
D’autres tubulopathies peuvent s’associer à une perte de sel et de potassium telles que les néphropathies interstitielles liées au syndrome de Sjögren, au lupus, à l’hypercalcémie et les lésions tubulaires induites par le lysozyme chez des patients atteints de leucémie aiguë myélomonocytaire.
On peut en rapprocher les syndromes polyuriques de toute cause. Normalement la concentration minimale de potassium urinaire est de 5 à 10 mmol/l. Lorsque le débit urinaire est de 5 à 10 litres par jour, il s’ensuit des pertes obligatoires de potassium de 50 à 100 mmol/jour. L’hypokaliémie survient plus généralement au cours des polydipsies primitives (souvent psychogéniques) au cours desquelles le débit urinaire peut être augmenté pendant des périodes de temps prolongées.
Tableau : Principales causes d’hypokaliémie Redistribution
Pertes digestives basses (diarrhée, laxatifs, tumeur villeuse) Pertes rénales
3. Traitement de l’hypokaliémie
Le traitement optimal de l’hypokaliémie dépend de la sévérité du déficit potassique mais aussi du mécanisme en cause : d’une façon générale, les suppléments potassiques sont indiqués lorsque l’hypokaliémie résulte d’un perte digestive ou rénale. A l’exception de la paralysie périodique hypokaliémique, les hypokaliémies de redistribution ne sont pas traitées par supplément potassique en raison du risque d’hyperkaliémie rebond, lorsque le processus initial est corrigé.
3.1. Déficit potassique
Il n’y a pas de corrélation stricte entre le déficit potassique et la concentration plasmatique de potassium. En général, la baisse de la kaliémie de 1 mmol/l correspond à une perte de 200 à 400 mmol de potassium. Pour des déplétions beaucoup plus importantes, la kaliémie baisse rarement en-dessous de 2,0 mmol/l.
Une acidose associée peut masquer l’iportance réelle de la déplétion potassique (par ex. acidocétose diabétique) L’hypokaliémie se démasque généralement lors de la correction de l’acidose et du déficit en insuline.
3.2. Préparation en potassium
D’une façon générale, le chlorure de potassium (KCl) doit être préféré pour corriger l’hypokaliémie. Celui-ci entraîne une augmentation plus importante de la concentration plasmatique de potassium que les sels alcalins. Par ailleurs, la plupart des patients ayant une hypokaliémie ont également une alcalose métabolique et l’administration de sels alcalins aggrave l’alcalose métabolique et agit comme anion non réabsorbable dans le rein favorisant l’excrétion urinaire de potassium.
Les sels alcalins de potassium, bicarbonate ou citrate, doivent être utilisés uniquement en cas d’acidose métabolique associée à l’hypokaliémie (acidose tubulaire rénale et diarrhées chroniques). Pour une raison similaire, l’apport d’aliments riches en potassium, (fruits frais, notamment oranges et bananes qui contiennent du phosphate et du citrate de potassium) est généralement moins efficace qu’un supplément en KCl.
3.3. Voie d’administration
La voie orale est souvent suffisante sous forme de suppléments oraux de KCl en préparation cristalline, liquide ou mieux tablettes à libération prolongée (Kaleorid®, Diffu-K®). Ces préparations à libération prolongée peuvent rarement causer des ulcérations de l’appareil digestif liées à l’accumulation locale de hautes concentrations de potassium.
L’administration IV de KCl est réservée aux patients incapables d’avaler ou avec une hypokaliémie sévère. 20 à 40 mmol de potassium sont dilués dans une solution salée de préférence [10] pour éviter les douleurs et la sclérose des veines périphériques. L’utilisation de doses plus importantes de potassium nécessite le recours à une veine centrale, en se méfiant des effets possibles d’une augmentation importante locale de la concentration de potassium sur la conduction cardiaque (en cas de voie centrale sous-clavière ou jugulaire).
3.4. Hypokaliémie légère à modérée
La plupart des patients ayant une hypokaliémie ont des valeurs entre 3 et 3,5 mmol/l. Ces hypokaliémies sont habituellement asymptomatiques, excepté chez les patients ayant une maladie cardiaque et traités par digitaliques ou une cirrhose hépatique avancée.
Le traitement consiste à remplacer les pertes de potassium par l’administration de 60 à 80 mmol de chlorure de potassium chaque jour.
Chez les patients sous diurétiques ou ayant un hyperaldostéronisme primitif, les pertes continues de potassium sont souvent associées à une hypomagnésémie qui doit être corrigée. Le recours à des diurétiques "épargneurs du potassium" (amiloride ou spironolactone) est généralement plus efficace chez ces patients car ces diurétiques réduisent à la fois la fuite urinaire de potassium et celle de magnésium. Les doses parfois importantes de spironolactone ou d’amiloride sont nécessaires compte-tenu des très hautes concentrations d’aldostérone observées dans ces situations. L’amiloride est généralement préféré en raison de l’absence d’effets secondaires gastro-intestinaux ou hormonaux (aménorrhée, gynécomastie) observés de façon dose-dépendante avec la spironolactone.
3.5. Hypokaliémie sevère ou avec signes ECG
En cas d’hypokaliémie sévère, il est nécessaire de corriger plus rapidement l’hypokaliémie chez les patients symptomatiques (arythmie, faiblesse musculaire). La voie orale reste tout à fait efficace et la concentration plasmatique de potassium peut monter de 1 à 1,5 mmol/l après 40 à 60 mmol et de 2,5 à 3,5 mmol/l après 135 à 160 mmol de potassium. Cet effet est souvent transitoire car une partie du potassium est captée par les cellules.
Chez certains patients ayant une hypokaliémie sévère, il est nécessaire de recourir à la voie IV. Une voie centrale est souvent préférable et en raison de l’utilisation de soluté salé, le risque de surcharge hydrosodée est un risque potentiel [11].
Une correction agressive par voie IV de l’hypokaliémie est nécessaire chez les patients en acidocétose diabétique ou en hyperglycémie sans cétose. Le traitement par insuline et la réhydratation exacerbent l’hypokaliémie dans ces situations.
En général la vitesse maximale d’administration du potassium par voie intraveineuse est 10 à 20 mmol/h (note) sous surveillance scopique permanente et rapprochée de la kaliémie [12].
[1] Une pseudohypokaliémie est exceptionnelle mais peut s’observer au cours de leucémies myéloïdes aiguës avec une hyperleucocytose importante (ces cellules métaboliquement actives peuvent capter le potassium après que le sang ait été prélevé). Dans ce cas, la concentration mesurée plasmatique de potassium peut être artificiellement abaissée de 1 mmol/l si le sang est conservé à la température ambiante.
[2] L’hypokaliémie stimule la réabsorption tubulaire proximale de bicarbonate et l’ammoniogénèse et en diminuant l’excrétion urinaire de citrate. L’hypokaliémie entraîne une acidification intracellulaire et inhibe la sécrétion d’aldostérone.
[3] L’hypokaliémie augmente l’ammoniogénèse dans le tube proximal et 50 % environ de cette production repasse dans la circulation systémique sous la forme d’ammoniaque. En cas d’insuffisance hépatique, la quantité d’ammoniaque est suffisante pour développer ou aggraver les symptômes d’encéphalopathie hépatique.
[4] Selon la dose et l’apport sodé, jusqu’à 50 % des patients traités par diurétiques, notamment thiazides de longue durée d’action, peuvent développer une hypokaliémie < 3,5 mmol/l. Malgré le bénéfice du traitement de l’hypertension artérielle, l’hypokaliémie induite par les diurétiques peut avoir des conséquences importantes en majorant le risque de mort subite chez les hypertendus traités par diurétiques thiazidiques en l’absence de diurétiques épargneurs du potassium.
[5] Ces kystes développés aux dépens de l’épithélium du tube collecteur sont souvent associés à des cicatrices interstitielles. La correction de l’hypokaliémie fait régresser les kystes. En augmentant l’ammoniogénèse et l’accumulation médullaire d’ammonium, l’hypokaliémie stimule l’activation du complément (formation de C3-amidé) générateur d’inflammation et de fibrose interstitielle.
[6] L’apport alimentaire normal en potassium est d’environ 1 mmol/kg/jour, davantage en cas d’alimentation riche en légumes et en fruits. L’adaptation rénale à la conservation du potassium permet d’excréter un minimum de 5 à 25 mmol/jour, même en présence d’une déplétion potassique sévère.
[7] Inversement la concentration en potassium du liquide gastrique est seulement de 5 à 10 mmol/l et la déplétion potassique dans ce contexte est essentiellement liée à une perte urinaire de potassium. En effet, la soustraction de liquide gastrique acide induit une alcalose métabolique et la fuite urinaire de potassium. Un élément diagnostique est la concentration abaissée de chlore urinaire dans ce contexte (voir "Alcalose métabolique").
[8] L’hypokaliémie secondaire à un abus de laxatifs peut être parfois difficile à mettre en évidence d’autant qu’elle s’associe parfois à un abus de diurétiques. La recherche de diurétiques dans les urines et une sigmoïdoscopie à la recherche d’une mélanose colique peut s’avérer utile chez les patients prenant des laxatifs de type anthracène pendant plus de 4 mois. En cas de laxatif à base de phénolphtaléine, l’alcalinisation des selles produit une couleur rose. Avec les catartiques contenant du magnésium ou du phosphate, la mesure directe de ces composés dans les selles permet de confirmer le diagnostic.
[9] La présence dans le tube collecteur cortical d’anions non réabsorbables majore le gradient électronégatif luminal et favorise la sécrétion de potassium. Dans ce cas, plupart du sodium délivré au tube distal est réabsorbé en échange de 1 potassium aboutissant à une sécrétion marquée de potassium. L’effet hypokaliémiant des anions non réabsorbables est beaucoup plus marqué lorsqu’il existe une déplétion volémique associée. Dans cette situation en effet, la réabsorption proximale augmentée diminue le débit distal de chlore délivré au tube collecteur et par ailleurs l’augmentation de sécrétion d’aldostérone favorise la sécrétion de potassium.
[10] Une solution à base de soluté salé doit être préférée aux solutions glucosées car celles-ci peuvent entraîner une diminution transitoire de la kaliémie de 0,2 à 1,4 mmol/l en raison de la stimulation de libération de l’insuline
[11] L’addition de potassium au soluté salé isotonique augmente considérablement l’hypertonicité de ces solutions et il est préférable d’ajouter le potassium à des solutions semi-isotoniques (soluté salé à 4,5 %o)
[12] à l’exception des sujets avec une paralysie périodique ou des arythmies menaçantes chez lesquels des doses de 140 à 100 mmol/h peuvent être nécessaires