|
|
|

|
|
OBJECTIFS
- Comprendre la diversité des présentaitons cliniques du lupus
- Evoquer le diagnostic et demander les sérologies de confirmation
|
|
|
 |
PLAN DU CHAPITRE
|
|
|
 Généralités Généralités
Le lupus érythémateux disséminé ou systémique (encore appellée maladie lupique) est une maladie inflammatoire chronique d'origine inconnue et qui affecte la peau, les articulations, le rein, les poumons, le système nerveux, les séreuses et/ou d'autres organes. Il est associé à des anomalies immunologiques spécifiques notamment la production de certains anticorps anti-nucléaires. L'évolution de la maladie lupique est caractérisée par des alternances de rémission et de rechute aigüe ou chronique.
Les femmes notamment dans la tranche 20 à 40 ans sont atteintes plus souvent que les hommes. La prévalence est de l'ordre de 5 à 250 cas pour 100 000 habitants, celle-ci étant beaucoup plus fréquente dans les populations antillaises, afro-américaine et hispano-américaine. Il existe un terrain génétique prédisposant ainsi qu'en témoigne le haut degré de concordance entre les jumeaux monozygotes et d'autre part l'association à des marqueurs génétiques comme HLA B8, DR2, DR3, DQW1, C2QO, C4QO, Gm, taux abaissé de CR1 et enfin un allèle de l'antagoniste du récepteur de IL-1. Quatre à huit gènes ont été impliqués dans la prédisposition au lupus. Des facteurs hormonaux notamment excès d'estrogènes actifs et/ou une diminution des androgènes expliquent la particulière fréquence de l'atteinte chez les femmes en période d'activité génitale et la possible induction ou exacerbation du lupus par les estrogènes thérapeutiques.
Les patients avec un lupus sont sujets à un très grand nombres de symptomes liés à l'atteinte inflammatoire qui peut toucher virtuellement tous les organes. La présentation la plus classique est une association de signes généraux et d'une atteinte cutanée, musculo-articulaire, hématologique et des modifications sérologiques.
Cependant, certains patients ont une atteinte prédominante hématologique, rénale ou neurologique. Le type de présentation prédominant au cours des premières années de la maladie tend à le rester par la suite.
L'atteinte rénale est cliniquement évidente chez environ 50 % des patients cependant la plupart sinon tous les patients restants ont une atteinte infraclinique qui peut être démontrée par la biopsie rénale. L'atteinte rénale survient habituellement pendant les premières années de la maladie et doit être détectée le plus précocément possible par une surveillance régulière biologique (protéinurie, sédiment urinaire, filtration glomérulaire). Plusieurs formes très différentes de glomérulonéphrite peuvent survenir au cours du lupus si bien que la biopsie rénale est indispensable pour caractériser le type et le degré de l'atteinte rénale. Le pronostic vital est bon si une thérapie appropriée est instituée suffisamment tôt.
Diagnostic et critères diagnostiques du lupus :
Le diagnostic du lupus est habituellement fait chez un patient se présentant avec 1 ou plusieurs des symptomes cliniques identifiés par l'American Rhumatologic Association (ARA). La présence d'une ou plusieurs anomalies sérologiques associées permet de conforter le diagnostic. Il est important cependant de réaliser que ces critères de lARA ont été développés pour la classification du lupus par comparaison à d'autres maladies rhumatologiques et avec pour objectif des études multicentriques. Ces critères de classification ont été établis par des analyses de cohortes et réalisées essentiellement dans des centres universitaires et chez des sujets de race blanche. L'extension de ces critères à d'autres populations n'a jamais été validée. Le diagnostic de lupus est retenu lorsque au moins quatre des manifestations sont présentes soit simultanément, soit séquentiellement (cf tableau critères de l'ARA). Lorsquils sont comparés avec les autres maladies rhumatologiques, ces critères de lupus ont une sensibilité, une spécificité d'environ 96 %
En pratique, il est important de reconnaître que de nombreux patients lupiques peuvent se présenter avec une atteinte mono-organique. D'autre part, un grand nombre de manifestations générales ne sont pas spécifiques. Pour faire un diagnostic précoce de lupus, il est donc nécessaire d'évoquer systématiquement ce diagnostic devant l'apparition isolée chez une femme jeune de fièvre, anémie, éruption photosensible, arthralgies ou arthrite, phénomène de Raynaud, polysérite, protéinurie ou syndrome néphrotique, symptome neurologique avec comitialité, alopécie, phlébite ou avortement récidivant.
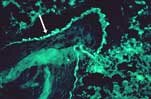
La démonstration d'un Coombs érythrocytaire positif, d'une hypocomplémentémie et de dépôts de complexes immuns à la jonction dermo-épidermique lors d'une biopsie cutanée en peau saine (lupus band test) sont également suggestifs de lupus.
La confirmation est généralement obtenue par la réalisation des anticorps anti-nucléaires.
Compte-tenu de la fréquence des lupus mono-organiques, une classification plus pratique peut être suggérée par analogie avec les critères de l'ARA pour le diagnostic de l'arthrite rhumatoïde :
- lupus défini : 4 ou plus de critères de l'ARA,
- lupus probable : 3 critères,
- lupus possible : 2 critères.
Parmi les patients classés en lupus probable ou possible, un certain nombre vont développer ultérieurement un lupus défini classiquement.
Utilisation, interprétation des auto-anticorps :
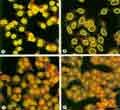
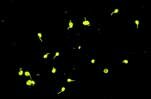
Les anticorps anti-nucléaires constituent le meilleur examen de dépistage du lupus et doivent être réalisés à chaque fois lorsque celui-ci est suspecté. Les anticorps anti-nucléaires sont positifs à titre significatif (habituellement au-dessus de 1/160ème ou plus) chez virtuellement tous les patients atteints de lupus bien qu'un anticorps anti-nucléaire positif est seulement une valeur prédictive de 20 à 35 % alors que la spécificité est pratiquement de 100 %, la probabilité d'avoir un lupus est inférieure à 0.14 % si les anticorps anti-nucléaires sont négatifs.
Des anticorps anti-nucléaires sont également présents mais habituellement à des titres plus faibles dans un grand nombre d'autres désordres rhumatologiques, les principaux étant : le syndrome de Sjögren (68 %), la sclérodermie (40 à 75 %), l'arthrite rhumatoïde juvénile (16 %), l'arthrite rhumatoïde (25 à 50 %).
Deux autres auto-anticorps sont eux hautement spécifiques du lupus, les anticorps anti-DNA natif (double brin) et les anticorps anti-Sm. Leur sensibilité est cependant plus faible, 75 et 25 % respectivement.
Certains auto-anticorps retrouvés au cours du lupus sont plus fréquemment retrouvés au cours d'autres maladies rhumatologiques associées à de anticorps anti-nucléaires avec une sensibilité, une spécificité variables :
- anticorps anti-DNA monobrin et anti-nucléoprotéine dans le lupus et l'arthrite rhumatoïde,
- anticorps anti-RNP dans le lupus et la sclérodermie,
- anticorps anti-Ro (ou anti-SSA) et anti-La (ou anti-SSB) dans le lupus et le syndrome de Sjögren.
Les associations suivantes ont été retrouvées entre certains anticorps, des manifestations plus spécifiques du lupus :
- anti-RNP (U1RNP) avec la myosite, le syndrome de Raynaud et un lupus moins sévère,
- anti-Ro avec lymphopénie, photosensibilité, lupus néonatal, déficit en C2 et syndrome de Sjögren,
- anti-SSB avec lupus néonatal et syndrome de Sjögren,
- anti-protéine P ribosomale avec psychose,
- anti-RA 33 avec l'arthrite érosive,
- anti-DNA natif avec la glomérulonéphrite,
- anti-Sm avec atteinte neurologique et glomérulonéphrite,
- les anticorps anti-C1q et anti-nucléosome sont fortement associés avec la survenue d'une glomérulonéphrite lupique et leur élévation précéde de 4 à 10 semaines environ d'aggravation ou la survenue clinique.
Pronostic :
L'évolution clinique du lupus est extrêmement variable depuis une maladie relativement bénigne jusqu'à une maladie rapidement progressive avec une atteinte mono-organique fulminante et le décès. La plupart des patients ont une évolution marquée par une alternance de poussées et de rémissions ces dernières étant probablement liées à l'utilisation de fortes doses de stéroïdes pendant les poussées les plus sévères.
La survie s'est considérablement améliorée au cours des dernières dizaines d'années depuis 40 % de survie à 5 ans dans les années 1950, actuellement 90 % à 10 ans. La probabilité de survie peut être évaluée sur la base du type d'organe atteint (les atteintes rénale et neurologiques étant les plus sévères) et le nombre de critères de l'ARA (plus celui-ci est élevé, plus le pronostic est mauvais). L'amélioration du taux de survie est probablement liée à de nombreux facteurs : meilleure reconnaissance de la maladie avec les tests sérologiques, diagnostic et traitement plus précoces, inclusion de cas plus bénins et meilleure utilisation des traitements et gestion de leurs complications.
La mortalité au cours du lupus est biphasique, la principale cause de mortalité dans les premières années de maladie est liée à l'activité de la maladie lupique elle même (atteintes rénale et neurologique en particulier) alors que les morts tardives sont essentiellement causées par les complications du traitement (notamment infections, insuffisance coronaire). Les infections sont favorisées par les glucocorticostéroïdes mais aussi les agents cytotoxiques. L'insuffisance coronaire est essentiellement liée à une athérosclérose accélérée qui est liée à l'utilisation des corticoides à forte dose. Par exemple l'augmentation de la dose quotidienne de prednisone de 10 mg est associée à une élévation de la cholestérolémie de 0.2 mmol/l. Le risque de survenue de cancer au cours du lupus est plus controversée. L'étude la plus importante chez 1585 patients danois atteints de lupus retrouvent une augmentation par 5 du risque de lymphome non hodgkinien et de cancer du poumon, du foie et du vagin.
La morbidité malgré la réduction de la mortalité à long terme, les patients lupiques ont un risque important de morbidité liée soit à l'activité de la maladie, soit aux effets secondaires des médicaments comme les corticostéroïdes ou les agents cytotoxiques. La nécrose aseptique de hanche est devenue un problème particulièrement fréquent et important compte-tenu de la prolongation de l'espérance de vie.
|
|
LECTURES RECOMMANDÉES
Mills JA. Systemic lupus erythematosus. New Engl J Med 1994;330:1871
Hahn BH. Antibodies to DNA. New Engl J Med 1998;338:1359
|
|
TEXTES COMPLÉMENTAIRES DANS LE SITE
Syndromes de néphropathie glomérulaire
Syndrome néphritique
Syndrome néphrotique
Glomérulonéphrites "primitives" ou "non spécifiques"
Lésions glomérulaire minimes
Hyalinose segmentaire et focale
Glomérulonéphrite extramembraneuse
Glomérulonéphrite endocapillaire aiguë post-infectieuse
Glomérulonéphrites membrano-prolifératives
Glomérulonéphrites "secondaires" ou "spécifiques"
Glomérulonéphrite à dépôts mésangiaux d'IgA,
Néphropathie diabétique,
Néphropathie lupique,
Amylose rénale,
Glomérulonéphrites extracapillaires (vascularites) associées aux ANCA,
Syndrome de Goodpasture,
Cryoglobulinémie
- Basalopathies héréditaires (
Syndrome d'Alport, Maladie des membranes basales minces)
|
|
Mise-à-jour Dim 17 sep 2000
Copyright © Tous Droits Réservés pour le site entier Nephrohus 2000
|
|
|
|
|