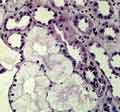
Ces éléments permettent déliminer les principaux diagnostics différentiels :
- les vomissements induits avec un chlore urinaire effondré et les lésions érosives caractéristiques des dents et des mains provoquées par lacide gastrique,
- labus et lintoxication par les diurétiques parfois plus difficile à éliminer sur la notion dune chlorurie variable dans le temps, augmentant sous diurétique et diminuant à leur arrêt. La certitude repose sur le dosage urinaire des diurétiques.
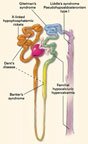
La similitude de présentation clinique entre les syndromes de Bartter et lintoxication ou labus de diurétiques a fait suggérer une anomalie génétique sur le ou les transporteurs du sodium classiquement inhibés par les diurétiques. Cette hypothèse est confirmée pour certaines formes de syndrome de Bartter et lun de ses variants cliniques appelé le syndrome de Gitelman.
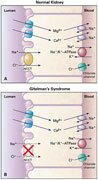
Le syndrome de Gitelman est caractérisé par une transmission autosomique récessive et les individus atteints sont des homozygotes composites (double hétérozygote) suggérant que la prévalence du gène n'est pas rare dans la population générale. Le gène codant pour NCCT dont la fonction est le co-transport NaCl dans le tube contourné distal, est le siège dune mutation inactivatrice. Plusieurs mutations ont été décrites et la plupart de ces mutations sont regroupées sur une portion commune de la chaîne carboxy-terminale intra-cellulaire. Cette mutation sur NCCT est responsable de la perte du transport vectoriel de sodium dans le tube contourné distal. La perte de chlorure de sodium entraîne une hypovolémie modérée avec activation du système rénine-angiotensine et lalcalose hypokaliémique. Il sy associe une augmentation de la réabsorption transcellulaire de calcium (lhyperpolarisation cellulaire semble stimuler la diffusion passive apicale de calcium ainsi que léchangeur sodium-calcium et la calcium-ATPase situés du coté basolatéral) si bien que la fuite urinaire de sodium saccompagne dune calciurie basse et quil ny a pas de néphrocalcinose.
Lhypomagnésémie constatée quasi constamment dans ce syndrome sexplique par une diminution de la réabsorption tubulaire de magnésium, celle-ci étant secondaire très probablement à la déplétion cellulaire en potassium.
Le syndrome de Gitelman est habituellement asymptomatique ou parfois responsable de signes neuro-musculaires (tétanie, spasmophilie) ou de chondrocalcinose.
Le syndrome de Bartter proprement dit est cliniquement différent du syndrome de Gitelman. Le syndrome de Bartter survient en effet plus volontiers dans la période néonatale, et se caractérise par des signes cliniques plus sévères : polyhydramnios, polyurie, polydipsie, déshydratation extra-cellulaire sévère et retard de croissance. Ce tableau saccompagne biologiquement dune alcalose hypokaliémique sévère, une magnésémie souvent normale (abaissée dans un tiers des cas), une calciurie élevée saccompagnant dans un très grand nombre de cas de néphrocalcinose et dinsuffisance rénale néonatale.
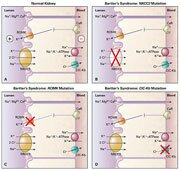
Le syndrome de Bartter dans sa forme néonatale reproduit les effets dune intoxication par les diurétiques de lanse si bien que lon a évoqué des anomalies (mutations inactivatrices) sur le gène codant pour NKCC2, protéine dont la fonction est le co-transport NaK2Cl assurant le transport vectoriel du sodium dans la branche ascendante large de lanse de Henle et inhibé par le bumétanide.
Une mutation inactivatrice sur NKCC2 a effectivement été retrouvée dans certaines familles exprimant une forme classique de syndrome de Bartter. Par contre des mutations inactivatrices ont été retrouvées sur au moins deux gènes codant pour deux autres transporteurs intervenants dans la réabsorption du sodium au niveau de lanse de Henle. Il sagit du gène codant pour ROMK, le canal potassique apical intervenant dans le recyclage du potassium et du gène codant pour ClCNKB un canal chlore baso-latéral de la cellule tubulaire de la branche ascendante large de lanse de Henle. Les mutations inactivatrices sur ces trois transporteurs sont responsables de phénotypes à peu près similaires avec un syndrome de Bartter classique sévère et dexpression néonatale.
Dans le phénotype du syndrome de Bartter classique, la perte de chlorure de sodium induit une hypovolémie sévère avec activation du système rénine-angiotensine et une alcalose hypokaliémique importante. La diminution de la réabsorption para-cellulaire du calcium (réabsorption passive le long du gradient électrique généré par NKCC2) est responsable de lhypercalciurie et de la néphrocalcinose. Labsence dhypermagnésurie et dhypomagnésémie semble liée à des mécanismes compensateurs dans les segments plus distaux du néphron.
La forme liée à linactivation de ClCNKB (Bartter de type III) nest cependant pas associée à la néphrocalcinose malgré lexistence dune calciurie modérément élevée. L'hétérogénéité génétique et phénotypique du syndrome de Bartter classique est récapitulée dans le Tableau 1 .
Sur le plan génétique, les individus affectés par le syndrome de Bartter classique sont presque toujours des homozygotes vrais avec la même mutation sur les deux gènes, traduisant le plus souvent une consanguinité. La prévalence du gène muté dans la population générale semble beaucoup plus rare que celle du gène responsable du Gitelman.
En pratique la distinction entre syndrome de Bartter et de Gitelman peut être assurée par le rapport calciurie/créatinine urinaire normal (>0,2 ) dans les syndromes de Bartter et abaissé (<0,2 et le plus souvent < 0,1) dans les syndromes de Gitelman. Ce rapport a un pouvoir discriminant de 100 % entre les deux syndromes.
Le mécanisme de lhypokaliémie, commun à ces anomalies génétiques (syndrome de Bartter, de Gitelman mais aussi syndrome de Liddle), se situe en regard de la cellule principale du tube collecteur cortical. Toutes ces hypokaliémies ont en commun un mécanisme faisant intervenir une augmentation de la sécrétion tubulaire apicale de potassium en échange de la réabsorption de sodium à ce niveau. Le facteur majorant lexcrétion tubulaire de potassium est lhyperpolarisation de la membrane apicale résultant de laugmentation de lélectronégativité luminale et traduisant soit une hyperactivité minéralocorticoïde soit une augmentation du débit luminal de sodium dans le tube collecteur cortical, soit enfin une réabsorption augmentée de sodium. Lensemble de ces mécanismes diversement associés est nécessaire pour majorer lexcrétion tubulaire de potassium et générer une hypokaliémie.
Sur le plan clinique, ces hypokaliémies tubulaires sont caractérisées par une kaliurèse > 20 mml/l inapropriée à lhypokaliémie et par un gradient transtubulaire du potassium (GTTK) supérieur à 5, témoignant dune activité minéralocorticoïde élevée. Lavantage du GTTK dans lévaluation dune hypokaliémie est que ce paramètre prend en compte la réabsorption deau dans le tube collecteur médullaire après le site de sécrétion du potassium et permet donc de différentier les situations où la kaliurèse est élevée en raison dune concentration des urines (réabsorption médullaire deau) et celles liées à une sécrétion distale inappropriée de potassium.
 3. Mutations affectant le canal sodium épithélial
3. Mutations affectant le canal sodium épithélial
Le syndrome de Liddle est cliniquement identifié depuis une trentaine dannées et se manifeste par une hypertension artérielle sévère souvent compliquée daccident vasculaire cérébral et associée à une hypokaliémie avec alcalose métabolique. Le caractère familial de cette affection a été reconnu depuis longtemps ainsi que le mode de transmission autosomique dominant. Classiquement, la présentation est précoce au cours de la vie, cependant les études génétiques ultérieures ont démontré une pénétrance variable en terme dhypertension artérielle et/ou dhypokaliémie.
Cette affection peut évoluer vers linsuffisance rénale terminale qui résulte de la sévérité de lhypertension, et/ou de lhypokaliémie chronique. Lhypertension artérielle est liée à lexpansion volémique qui explique aussi la rénine effondrée, laldostéronémie effondrée et peu stimulable et une cortisolémie normale.
La spironolactone ainsi que ladrénalectomie sont inefficaces dans cette affection. Par contre, lamiloride à dose souvent élevée (10 à 30 mg/jour) ou le triamtérène sont efficaces. A noter également que la transplantation rénale corrige lensemble des anomalies (y compris une réponse normale de la rénine et de l'aldostérone à la déplétion sodée et à l'orthostatisme) suggérant que lhypertension et lhypokaliémie sont liées à une anomalie rénale.
Lanomalie génétique sous-tendant le syndrome de Liddle a été récemment mise en évidence. Il sagit dune mutation sur la séquence carboxy-terminale intra-cellulaire de la sous-unité béta ou de la sous-unité gamma du canal épithélial sodique ENaC, présent dans la membrane apicale des cellules principales du tube collecteur cortical. Cette mutation est responsable dune activation constitutionnelle du canal sodium et ceci favorise laugmentation de la réabsorption sodée. Cette dernière est responsable de lexpansion volémique, de lhypertension artérielle et de la suppression du système rénine-angiotensine, avec atrophie de lappareil juxta-glomérulaire. Laugmentation de lélectro-négativité luminale est responsable de laugmentation de lexcrétion de potassium.
Les mécanismes cellulaires liant la mutation et lactivation constitutionnelle du canal sont encore controversés. La conséquence fonctionnelle est laugmentation de probabilité douverture du canal qui traduit une augmentation du nombre des canaux exprimés à la surface cellulaire, résultant elle-même d'une diminution du catabolisme intracellulaire des canaux. Labsence de régulation négative du canal sodium épithélial face à lexpansion volémique persistante sous-tend le mécanisme physiopathologique de ce syndrome.
Cette anomalie originale de mutation activatrice constitutionnelle du canal ENaC a fait suggérer quune inactivation de ce même canal pourrait avoir des conséquences cliniques spécifiques.
Des mutations inactivatrices ont été identifiées et sont responsables dun syndrome clinique identifié depuis longtemps comme le pseudo-hypoaldostéronisme de type 1. Cette affection rare qui se transmet sur un mode autosomique récessif est responsable dun tableau néonatal sévère avec perte de sel, hypovolémie, hypotension, hyperactivation marquée du système rénine-angiotensine, acidose métabolique hyperkaliémique et des infections pulmonaires récurrentes simulant une mucoviscidose. Cette affection est liée à une mutation inactivatrice du canal sodique épithélial, la mutation pouvant survenir sur la chaîne alpha, béta ou gamma de ce canal. Cette mutation est responsable dune diminution de la probabilité douverture du canal ENaC, dune résistance généralisée à laction de laldostérone et d'un déficit permanent atteignant tous les organes cibles de laldostérone (reins, côlon, glandes salivaires ainsi que les poumons). Cette affection est donc caractérisée sur le plan phénotype par un syndrome clinique en miroir de celui du syndrome de Liddle.
Le PHA1 autosomique récessif doit être distingué de la forme autosomique dominante caractérisée par un tableau proche de perte de sel mais sans atteinte pulmonaire ou dautre organes. Le PHA1 dominant est lié à une mutation sur le gène codant pour le récepteur aux minéralocorticoïdes (MR). La carbenexolone corrige partiellement la résistance apparente aux minéralocorticoïdes. En ralentissant la conversion de cortisol en cortisone la carbenoxolone augmente la concentration intracellulaire de cortisol suffisamment pour maintenir lactivité du récepteur "sauvage" prenant le pas sur le défaut fonctionnel du récepteur mutant inactif (haplo-insuffisance).
4. Mutations affectant le récepteur-senseur du calcium extracellulaire
Un syndrome d'hypercalcémie familiale bénigne est connu depuis plusieurs décennies. Cette affection est caractérisée aussi par une hypocalciurie et a été également appelée hypercalcémie familiale hypocalciurique. Les patients ont une hypermagnésémie modérée, une concentration d'hormone parathyroïdienne normale ou légèrement augmentée. Les études de charge calcique montrent une augmentation du seuil de libération de l'hormone parathyroïdienne. La persistance de l'hypercalcémie et l'hypocalciurie après parathyroïdectomie indiquent que l'hyperparathyroïdie n'est pas le mécanisme primitif de cette affection et que le seuil de régulation rénal est également altéré.
Chez certains enfants issus de parents consanguins avec une hypercalcémie familiale hypocalciurique, une hyperparathyroïdie sévère caractérisée par une hypercalcémie, un retard de croissance, une ostéopathie et des fractures multiples se développent rapidement après la naissance.
Les études de clonage ont identifié le récepteur-senseur du calcium extra-cellulaire qui est un récepteur de 1078 aminés, couplé à une G-protéine. Dans le rein, le récepteur-senseur du calcium est exprimé à la surface baso-latérale des cellules de la branche ascendante large de l'anse de Henle, sur la surface luminale des cellules du tube collecteur papillaire et dans d'autres segments du néphron. Ces sites sont ceux où l'hypercalcémie est connue pour inhiber la réabsorption du chlorure de sodium, du calcium et du magnésium au niveau de l'anse ascendante large de Henle et pour inhiber l'effet hydro-osmotique de la vasopressine dans le tube collecteur.
Des mutations du gène du récepteur-senseur du calcium ont été mis en évidence, ce gène étant situé sur le chromosome 3. Plusieurs dizaines de mutations différentes ont été maintenant identifiées chez ces patients. Environ deux tiers des familles atteintes ont une unique mutation hétérozygote du récepteur-senseur du calcium. Ces mutations modifient le fonctionnement du récepteur-senseur du calcium qui ne répond qu'à des concentrations de calcium supérieures à la normale ce qui correspond à un déplacement vers la droite du seuil de régulation des réponses calcium-dépendantes. L'hyperparathyroïdie néonatale sévère est habituellement liée à des mutations homozygotes chez des enfants dont les parents sont consanguins avec une hypercalcémie hypocalciurique.
Des mutations du gène s'accompagnant de gain de fonction du récepteur censeur du calcium induisent une hypocalcémie avec une concentration normale d'hormone parathyroïdienne. Le récepteur répond à des concentrations de calcium plus basses que normales. Les patients avec ce type de mutation ont aussi une hypercalciurie et certains une néphrocalcinose et une atteinte rénale suggérant que des anomalies du récepteur-senseur du calcium pourraient contribuer à certaines formes d'hypercalciurie idiopathique.