|
|
|
|

|
|
OBJECTIFS
|
|
|
 |
PLAN DU CHAPITRE
|
|
|
Le syndrome d'Alport est une forme de néphropathie glomérulaire héréditaire progressive, du moins chez les sujets masculins, et qui est souvent associée à une surdité et à des anomalies lenticulaires.
 1. Génétique 1. Génétique
Dans la plupart des familles, ce désordre est transmis selon un mode lié à l'X. Ceci signifie quil ny a pas de transmission du père à son fils et que les femmes sont porteuses de l'X anormal avec grossièrement 50 % des cellules atteintes (inactivation aléatoire de X selon les cellules d'après l'hypothèse de Marie Lyon). Ces femmes sont porteuses mais généralement peu ou pas symptomatiques.
Plus rarement un mode de transmission autosomique récessif est en cause. Dans cette forme, les femmes sont atteintes aussi sévèrement que les hommes et/ou la transmission de père à fils peut survenir. Les manifestations cliniques sont virtuellement identiques à celles du syndrome d'Alport liées à l'X classique mais le défaut génétique en cause concerne une chaîne du collagène différente.
2. Physiopathologie
L'anomalie primitive chez presque tous les patients avec un syndrome d'Alport réside dans le domaine non collagène (NC1 du collagène du type 4) et correspond à une anomalie du gêne codant pour la chaîne alpha 5 du collagène 4, gène situé sur le chromosome X (gène COL 4 A 5). L'antigène de Goodpasture est un épitope de la chaîne alpha 3 du collagène du type 4 dont le gène est situé sur le chromosome 2. Les chaînes alpha 3 et alpha 5 ont une distribution analogue dans le glomérule et ces deux chaînes se combinent pour former un type particulier de collagène. Une anomalie de la chaîne alpha 5 peut donc limiter la formation de ce nouvel antigène et prévenir l'incorporation des chaînes alpha 3 et alpha 4 contenant l'antigène de Goodpasture.
Le syndrome d'Alport à transmission autosomique récessive correspond à des anomalies sur la chaîne alpha 3 ou alpha 4 dont les gènes sont codés tête à tête sur le chromosome 2. Une anomalie de l'une de ces chaînes altère l'intégrité de la membrane basale glomérulaire et de la cochlée entraînant des manifestations cliniques similaires au syndrome d'Alport classique. Différentes mutations de la chaîne alpha 4 et peut-être alpha 3 sont responsables de certains cas de maladies des membranes basales minces, une affection transmise selon un mode autosomique dominant mais qui ne progresse habituellement pas vers l'insuffisance rénale.
La chaîne alpha 5 est exprimée essentiellement dans la membrane basale glomérulaire rénale et les mutations survenant à différents sites du gène de la chaîne alpha 5 altèrent la structure de la membrane basale. Dans le syndrome d'Alport lié à l'X , la membrane basale glomérulaire est caractérisée par l'absence de chaînes alpha 3, alpha 4 et alpha 5 et par la persistance d'une distribution foetale de chaînes alpha 1 et alpha 2. Il semble que les chaînes alpha 3, alpha 4 et alpha 5 normalement riches en cystéine augmentent la résistance de la membrane basale glomérulaire vis-à-vis des agressions protéolytiques au niveau du site de filtration. La persistance des isoformes foetales confère une susceptibilité accrue à l'attaque protéolytique par les collagénases et les catepsines. Ces agressions protéolytiques sur des périodes prolongées pourraient être responsables de la fragmentation de la membrane basale, éventuellement de la glomérulosclérose.. Les anomalies de fonction du collagène sont également responsables des manifestations extrarénales du syndrome d'Alport (cochlée et lenticone).
3. Manifestations cliniques
Les manifestations rénales initiales du syndrome d'Alport sont une protéinurie et/ou une hématurie asymptomatique ou encore des épisodes récidivant d'hématurie macroscopique. L'hématurie commence avant l'âge de 5 ans chez les garçons mais peut ne pas être diagnostiquée jusqu'à l'âge adulte. La concentration plasmatique de créatinine, la pression artérielle sont normales initialement mais une insuffisance rénale progressive, une hypertension artérielle, une protéinurie évoluant vers le syndrome néphrotique surviennent avec le temps.
L'insuffisance rénale terminale survient généralement chez les garçons entre l'âge de 16 et 35 ans mais l'évolution peut être parfois plus lente dans certaines familles avec une insuffisance rénale retardée jusqu'à l'âge de 45 à 60 ans. La variabilité clinique pourrait résulter en partie de mutations différentes du gène alpha 5 : les sujets de sexe masculin infectés et ayant une délétion complète du gène pourraient avoir une maladie plus sévère à la fois rénale et extrarénale que ceux atteints par une mutation ponctuelle du gène.
Les anomalies histologiques rénales augmentent de sévérité avec l'âge. Les manifestations les plus précoces consistent en un amincissement de la membrane basale glomérulaire. Cet aspect n'est pas pathognomonique et peut survenir dans d'autres affections dont notamment la maladie des membranes basales minces. Dans le syndrome d'Alport cependant, l'évolution se fait vers le développement de fragmentation longitudinale de la membrane basale produisant un aspect laminé et feuilleté. Chez les sujets masculins, le nombre de glomérules atteints de ces fragmentations augmente de 30 % à l'âge de 10 ans jusqu'à plus de 90 % à l'âge de 30 ans. Inversement moins de 30 % des glomérules sont touchés chez la plupart des femmes qui ont peu ou pas d'atteinte glomérulaire. Les modifications en microscopie optique sont moins spécifiques et comportent des augmentations focales de la cellularité du glomérule progressant vers la glomérulosclérose et un infiltrat interstitiel riche en lipophages.
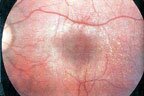
La manifestation extrarénale la plus fréquente est une perte de l'audition (surdité de perception) qui commence dans les fréquences élevées et progresse vers la surdité. Les manifestations oculaires comprennent lenticone antérieur, cataracte et des lésions périmaculaires blanchâtres de la rétine. Ces lésions maculaires sont toujours tardives et concurrente d'une insuffisance rénale. Une dysfonction plaquettaire avec plaquettes géantes est présente dans quelques familles avec une forme autosomique récessive.
4. Diagnostic
Le diagnostic de syndrome d'Alport est habituellement suspecté sur la notion d'une histoire familiale avec insuffisance rénale et surdité. Ceci permet de distinguer cette affection des autres causes d'hématurie glomérulaire récidivante : la néphropathie à IgA au cours de laquelle l'histoire familiale est habituellement négative et la maladie des membranes basales minces au cours de laquelle il existe une histoire familiale mais où l'insuffisance rénale et la surdité sont typiquement absentes. Dans 15 % des cas environ, on ne retrouve pas d'histoire familiale d'atteinte rénale et le diagnostic est établi sur la biopsie rénale. Ces patients représentent probablement de nouvelles mutations du gène responsables des anomalies des membranes basales.
Il est possible de différencier certaines de ces affections à partir d'échantillons de biopsie cutanée et l'utilisation d'un anticorps monoclonal dirigé contre l'antigène de la chaîne alpha 5.
Les hommes atteints de syndrome d'Alport ont une absence complète de fixation le long des membranes basales épidermiques alors que les femmes porteuses asymptomatiques ont une fixation discontinue. Les sujets ayant une maladie des membranes basales minces ont une fixation normale. Les analyses génétiques directes du gène COL 4 A 5 par PCR et séquençage de l'ADN pourraient s'avérer des techniques diagnostiques de choix dans l'avenir.
5. Traitement
Il n'y a pas de traitement du syndrome d'Alport. Un traitement par inhibiteur de l'enzyme de conversion permet cependant de réduire la protéinurie au moins chez certains patients, de même que la vitesse de progression de l'atteinte rénale. Ce type de traitement doit donc être proposé à tout patient atteint de syndrome d'Alport, hypertendu ou non.
Au stade d'insuffisance rénale terminale, le traitement fait appel à la dialyse ou à la transplantation. La maladie ne récidive pas sur le transplant mais 3 à 4 % des patients développent une maladie des anticorps anti-membrane basale glomérulaire de novo. Les anticorps sont généralement dirigés contre l'antigène de Goodpasture, situé dans la chaîne alpha 3, et sont similaires aux anticorps présents dans les formes "classiques" de maladie anti-GBM. Cet antigène est reconnu comme étranger dans le greffon, probablement parce qu'il n'est pas exprimé dans le rein natif en raison de l'absence de formation du collagène spécifique contenant l'association de chaînes alpha 3- alpha 5.
La maladie des anticorps anti-GBM survient généralement au cours de la première année de transplantation. Plus de 75 % des patients ayant des anticorps anti-GBM circulants développent une glomérulonéphrite extracapillaire avec le risque de perte du transplant. Les traitements à base d'échanges plasmatiques et de cyclophosphamide utilisés au cours des maladies à anti-GBM primitives apparaissent de bénéfice très limité dans ce contexte particulier. La retransplantation est associée à un risque élevé de récidive. Il est possible que le type du défaut génétique dans le gène de la chaîne alpha 5 joue un rôle important dans la génèse de ces anticorps anti-GBM posttransplantation. Le risque apparaît plus important chez les patients ayant de grandes délétions géniques chez lesquels l'épitope de Goodpasture sur la chaîne alpha 3 pourrait ne pas être exprimé.
Il est important de souligner que l'incidence relativement faible de l'anticorps anti-GBM dans ce contexte signifie que globalement la transplantation rénale n'est pas contre-indiquée chez les patients avec un syndrome d'Alport. Cependant le traitement et l'attitude optimale ne sont pas bien définis chez les patients qui ont déjà perdu une fois leur transplant en raison de l'apparition d'anticorps anti-GBM. L'incidence des récidives est élevée dans ce contexte, proche de 90 %.
|
|
LECTURES RECOMMANDÉES
|
|
TEXTES COMPLÉMENTAIRES DANS LE SITE
|
|
Mise-à-jour : vendredi 5 mai 2000
Copyright © Tous Droits Réservés pour le site entier Nephrohus 2000
|
|
|
|