|
 1. Présentation clinique 1. Présentation clinique
Le syndrome néphrotique à lésions glomérulaires minimes représente 80 à 85 % des syndromes néphrotiques idiopathiques observés chez lenfant avec une prévalence pic entre lâge de 4 et 8 ans. Environ 20 % des adultes avec un syndrome néphrotique idiopathique peuvent également présenter ce type de lésions. Les hommes sont affectés légèrement plus souvent que les femmes.
Le début de la maladie est habituellement brutal sous la forme dun oedème facial ou périorbital ou une tension abdominale.
Dans certains cas les oedèmes sont massifs avec ascite et épanchement pleural (polysérite) parfois responsable de détresse respiratoire. Une infection révélatrice est moins fréquente et revet la forme dune péritonite primitive, dune septicémie ou dune cellulite infectieuse. Presque toutes ces infections (sans porte dentrée évidente) sont causées par un Streptococcus pneumoniae ou un germe entérique.
Presque tous les patients se présentent avec un syndrome néphrotique et une fonction rénale relativement bien préservée. Cependant une forme réversible dinsuffisance rénale aiguë (fonctionnelle ou nécrose tubulaire aiguë) peut être exceptionnellement observée, celle-ci étant en rapport avec lhypovolémie sévère observée au cours de ce type de syndrome néphrotique.
Le sédiment urinaire est classiquement bénin mais dans environ 20 % des cas une hématurie microscopique de faible abondance peut être présente.
Lhypertension est classiquement absente. La protéinurie est par définition supérieure à 3 g/j (adulte) ou 50 mg/kg/j (enfant), mais souvent très abondante 6 à 10 g/j avec une hypoalbuminémie profonde (<15-20 g/l). La protéinurie est dite sélective car composée en grande majorité dalbumine et pas ou peu dimmunoglobuline.
Les concentrations sanguines de cholestérol et de triglycérides sont élevées souvent de façon importante. La concentration sérique de complément et de ses fractions (C3, C4) sont classiquement normales. On retrouve souvent une incidence augmentée de terrain atopique et un excès de fréquence dantigène HLA B8, B13 et DR7.
2. Diagnostic
Le diagnostic de LGM repose sur la biopsie rénale systématique chez ladulte et le grand-enfant en raison de la grande fréaquence des diagnostics différentiels à cet âge. Chez lenfant entre 4 et 8 ans la biopsie rénale nest habituellement pas préconisée en raison de la très forte prévalence de la LGM dans cette tranche dâge et une corticothérapie empirique est instituée. Par contre la biopsie rénale est indiquée demblée en cas de présentation clinique atypique ou secondairement en cas de résistance au traitement glucocorticoide.
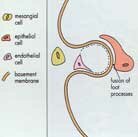
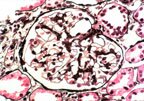
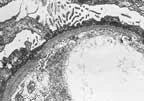
Le syndrome néphrotique à lésions glomérulaires minimes est caractérisé par des glomérules normaux ou très modérément altérés en microscopie optique, labsence de dépôts dimmunoglobuline en immunofluorescence et des lésions épithéliales diffuses avec effacement ou fusion des pédicelles en microscopie électronique. Les dépôts électron-denses sont habituellement absents mais peuvent être exceptionnellement observés en petite quantité dans le mésangium. Une hypercellularité mésangiale très minime, des anomalies tubulaires focales et occasionnellement un glomérule sclérotique globalement sont également compatibles avec ce diagnostic. Si léchantillon de tissu rénal est de petite taille comportant moins de 10 glomérules, des lésions de hyalinose segmentaire et focale peuvent facilement être manquées en particulier si la biopsie a intéressé la corticale superficielle (voir « hyalinose segmentaire et focale »).
3. Causes ou associations
Des lésions rénales similaires à la LGM peuvent être observées en association avec certaines maladies, notamment les hémopathies comme la maladie de Hodgkin ou dautres formes de lymphomes, certains cancers (cancer du rein, de la prostate, mésothélium, cancer du pancréas), des allergies au pollen ou au lait, les piqures de guêpe ou dortie ou encore des traitements par certains médicaments.
Les anti-inflammatoires non stéroïdiens, en particulier le fénoprofène, représentent une des causes les plus fréquentes de LGM secondaire. La plupart mais pas tous les patients développent parallèlement une néphropathie interstitielle aiguë avec insuffisance rénale, hématurie et pyurie. Une présentation similaire peut être observée avec lampicilline, la rifampicine et linterféron. Larrêt du médicament en cause entraîne la résolution du syndrome néphrotique. Les sels dor, le lithium ou la thiopronine ont également été incriminé à lorigine de LGM mais ces médicaments sont plus habituellement responsables de glomérulonéphrite extra-membraneuse et celle-ci peut être parfois méconnue en labsence dexamen histologique attentif en microscopie électronique.
En cas de LGM secondaire à une hémopathie, lactivité de latteinte rénale est généralement parallèle à celle de la maladie hématologique et la protéinurie disparait lors de la rémission induite par la chimiothérapie. Dans pratiquement tous les cas lhémopathie sous-jacente est déjà soit diagnostiquée soit cliniquement apparente lors de lapparition du syndrome néphrotique. Une évaluation extensive à la recherche dune néoplasie occulte nest donc pas indiquée chez les patients avec une LGM en labsence de signes évocateurs comme une perte de poids, une fièvre inexpliquée, des adénopathies. De même la recherche de tumeur ne doit pas être entreprise sur la seule notion dune vitesse de sédimentation élevée, celle-ci étant souvent augmentée jusquà 100 mm/h au cours du syndrome néphrotique.
La pathophysiopathologie du syndrome néphrotique à lésions glomérulaires minimes reste inconnue. Aucune immunoglobuline ne semble impliquée. Plusieurs éléments suggèrent un mécanisme immunologique : association avec des maladies infectieuses ou une immunothérapie prophylactique (vaccins) ; association avec un terrain atopique ; prévalence augmentée en association avec certains antigènes HLA ; association à une anomalie de fonctionnement des cellules T (maladie de Hodgkin) ; réponse à la corticothérapie et rémission lors de maladies iontercurrente comme la rougeole ; sécrétion de lymphokine augmentant la perméabilité capillaire.
Lurine contient principalement de lalbumine protéine fortement anionique, ce qui résulte dun défaut spécifique sur la barrière de charge anionique de la paroi capillaire alors que la permsélectivité de taille semble relativement bien préservée (voir « permsélectivité glomérulaire »).
Tableau 1 : Principales causes de LGM
- Idiopathiques (presque tous les cas)
- Médicaments (AINS, ampicilline, rifampicine, interféron)
- Hémopathies (Hodgkin, lymphomes, leucémies)
- Certains cancers solides ( ?)
- Allergies (allergènes alimentaires, piqures de guêpe, pollens etc&)
|
4. Evolution et traitement
Le syndrome néphrotique à lésions glomérulaires minimes non compliqué par une hyalinose segmentaire et focale surajoutée ou une hypercellularité mésangiale sévère est globalement une maladie relativement bénigne. Les rémissions spontanées peuvent survenir dans 30 % des cas environ. Moins de 5 % des patients progressent vers linsuffisance rénale terminale, ceci toujours après une évolution (ou lexistence demblée) de lésions de hyalinose segmentaire et focale. Cependant, en raison des manifestations invalidantes du syndrome néphrotique et de laugmentation nette de la susceptibilité aux accidents thromboemboliques et aux infections, la plupart des enfants et des jeunes adultes sont traités.
Les glucocorticoïdes induisent un taux important de rémission.
- Plus de 90 % des enfants développent une rémission complète avec 60 mg/m2/jour ou 1 mg/kg de prednisone ou de prednisolone orale (80 mg/jour maximum) pendant une durée de 4 à 6 semaines suivie par 40 mg/m2 tous les deux jours pendant 4 à 6 semaines.
Chez les non-répondeurs, il existe habituellement une maladie sous-jacente non reconnue sur la biopsie rénale initiale, notamment une hyalinose segmentaire et focale. Les sujets répondeurs ont tendance à rechuter lorsque les stéroïdes sont arrêtés ou lors dune diminution de la posologie de stéroïdes, notamment si la diminution est trop rapide. Globalement, environ 60 % des patients développent au moins un épisode de rechute. Ces patients avec rechute fréquente ou dépendant des stéroïdes nécessitent généralement un traitement plus prolongé. Des rechutes moins fréquentes ou très espacées peuvent être traitées par des courses répétées de glucocorticoïdes sauf si une toxicité sévère des stéroïdes survient.
- Les adultes nécessitent un traitement plus prolongé de prednisone ou de prednisolone 1 mg/kg/jour (maximum 80 mg/jour) pendant 8 à 10 semaines suivi par une décroissance progressive sur quelques mois. Le taux de rémission globale est à peu près équivalent chez ladulte et chez lenfant cad 80 à 85 %. La taux de rechute est plus important chez ladulte (65-80%) dont 70% survenant dans les 3 premiers mois suivant une rémission.
Une dépendance vis-à-vis des stéroïdes ou des récidives très fréquentes peuvent également être traitées avec succès par des agents alkylants aussi bien chez lenfant que chez ladulte. Le cyclophosphamide oral 2 à 3 mg/kg/jour pendant 10 à 12 semaines semble également efficace et relativement bien toléré. La dose cumulative de cyclophosphamide ne doit cependant pas dépasser 200 mg/kg. La rémission doit initialement être obtenue avec les stéroïdes lorsque cela est possible. Si une rechute survient, un traitement par cyclophosphamide peut être tenté. Environ 50 à 60 % des patients traités de cette façon sont mis en rémission pendant une période prolongée.
La cyclosporine sans dépasser 5 mg/kg/jour, associée à des faibles doses de glucocorticoïdes peut être utilisée comme une alternative au traitement par cyclophosphamide ou encore chez les patients qui ne répondent pas au cyclophosphamide ou qui récidivent rapidement à larrêt de celui-ci. Malheureusement, les récidives sont fréquentes lorsque la cyclosporine est arrêtée (cyclosporine-dépendance). Globalement 70 à 75 % des patients répondent au traitement par cyclosporine mais ce pourcentage est franchement moindre pour les malades résistant aux corticoïdes qui sont justement ceux chez lesquels la cyclosporine peut être indiquée (85-90% des répondeurs aux stéroides, 60-70% des résistants aux stéroides). Labsence de réponse à la cyclosporine après 3 mois de traitement, traduit habituellement une maladie réfractaire de mauvais pronostic fonctionnele rénal à long terme. Si une rémission peut être induite par des doses de cyclosporine de 5 mg/kg ou moins, une dose habituellement plus faible peut être maintenue pendant 12 mois avant dessayer de décroître ou darréter le traitement.
Plus rarement le lévamisole (enfant) ou lazathioprine (adulte) peuvent être utilisés pour induire ou maintenir une rémission en cas de forme multi-récidivante mais ces produits sont insuffisamment validés.
Globalement lévolution à long terme de la LGM cortico-sensible est favorable et très peu de patients développent une insuffisance rénale. La plupart des décès sont liés aux infections, aux thromboembolies ou aux complications du traitement.
5. Variant clinique de la LGM : néphropathie à C1q
La néphropathie à C1q est un variant de la LGM et correspond à une affection mal définie mais comportant une prolifération mésangiale, des dépots mésangiaux en microscopie électronique, dépots essentiellement composés de C1q en IF. Des dépots très modérés d IgG, IgA et IgM peuvent également être observés suggérant un mécanisme à complexes immuns.
Comme la néphropathie à IgM, la néphropathie à C1q peut se présenter par une hématurie, une protéinurie asymptomatique ou un syndrome néphrotique. Les sujets jeunes mâles noirs paraissent plus communément atteints.
Une rémission complète après corticothérapie est inhabituelle et la plupart des patients sont soit cortico-résistants soit cortico-dépendants. Le bénéfice du cyclophosphamide nest pas établi. Le risque de progression vers linsuffisance rénale est plus important en cas de protéinurie abondante persistante et délévation initiale de la créatininémie.
|